
Specific regulations/requirements of International Markets
11 january 2024
Regulatory affairs refer to the processes and activities involved in ensuring that products, whether pharmaceuticals, medical devices, cosmetics, food, or other regulated items, comply with the relevant regulations and laws in a given country or region. The specifics of regulatory affairs can vary significantly from one country or region to another due to differences in regulatory bodies, laws, and standards. Thus, the pharmaceutical market is divided into two categories: a regulated market and an International market.(1)
Regulated market:
A regulated market is controlled and defined by well-structured guidelines that guarantee the safety and quality of the drugs and their products. The United States, the European Union (EU), and Japan form the regulated market. Each region has a well-structured agency; for example, the European Medicines Agency (EMA) guarantees the EU countries get the drug’s marketing authorization or the final product through a cascade of well-defined procedures.(2)
International market:
The regulations in the international markets are still far from defined, and each country has its requirements. The international markets’ socio-economic growth potential makes them attractive, so pharmaceutical companies must keep up-to-date with the latest regulatory developments to ensure their place in these markets.
Product registration in an international market is quite challenging as they are not harmonized and rich in diversity of regulatory requirements.
The international, sometimes referred to as the semi-regulated market, comprises Asia Pacific, Eastern Europe, Africa LATAM, and Gulf countries.
Here is a little description of each region’s requirements and measures to harmonize its pharmaceutical regulations.(3)
Africa
The researchers discovered that over 85% of sub-Saharan Africa is currently engaged in initiatives to harmonize the registration of medicines. However, challenges have arisen due to issues related to substandard drugs. This is primarily because most pharmaceutical products are imported to the continent, and the effective enforcement of regulations in certain countries has proven challenging.(4)
The African Union has opted for the African Medicines Agency (AMA) to harmonize the regulatory system. The AMA will be crucial in ensuring medicinal products’ safety, efficacy, and quality, plus a global harmonization meeting international standards.(5)
Many African nations have adopted the Common Technical Document (CTD) format as their primary submission format for regulatory purposes. However, some countries, like Sudan, continue using their unique submission formats. In contrast, others are open to receiving submissions in either the CTD format or a specific format of their choice.
Inspirations for regulatory systems in certain African countries are drawn from well-established models, such as France’s ANSM. For instance, France’s regulatory framework influences most Northern and French-speaking African nations and predominantly follows the CTD model.
In contrast, countries like South Africa have implemented the electronic Common Technical Document (eCTD) format for submitting regulatory events.
Asia Pacific
As for Asia, the spotlights are directed towards the Association of Southeast Asian Nations (ASEAN); the member countries are Brunei Darussalam, Cambodia, Indonesia, Lao PDR (Laos), Malaysia, Myanmar (Burma), Philippines, Singapore, Thailand and Vietnam.
The ASEAN region has developed ASEAN Common Technical Requirements (ACTRs), which provide guidelines for compiling drug dossiers and are comparable to Notice to Applicants (NTA) Volume 2C in Europe.(6)
- ACTD format
The ACTD format differs slightly from the ICH CTD format used in Europe. The ACTD consists of Parts I to IV, whereas ICH CTD has 5 Modules. Parts II to IV correspond to Modules 3 to 5, respectively. In ACTD, the summaries of the quality (Part II), nonclinical (Part III), and clinical (Part IV) are at the beginning of each part of the ACTD sections. In eCTD, there is a specific module 2 for these same summaries.(7)
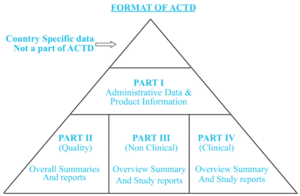
Figure 1: ACTD format
The ACTD is only accepted for generic applications in Indonesia, Vietnam, Singapore, Philippines, Malaysia, and Thailand, whereas the CTD format is acceptable for innovator, biological, and biotechnological products.
Only Thailand accepts the eCTD format, while Singapore and Malaysia accept electronic submissions in CTD format through their online system. In the Philippines, submission is done mainly via email, while digitalization of the submission portal is underway to accommodate online dossier submissions. In some instances, physical paper and DVD submissions are still required in certain countries.(8)
Common requirements for registration:
To register a drug product in the ASEAN region, several requirements need to be met:
- Site Registration: It is mandatory to undergo site registration.
- Plant GMP Approval: The ASEAN region accepts FDA/EU/PICs (Pharmaceutical Inspection Cooperation Scheme) approval for the finished product site.
- Stability Zone: ASEAN countries fall under stability zones IVa and IVb. The stability conditions for these zones are as follows:
- Zone IVa: 30±2°C/65±5% RH (Relative Humidity).
- Zone IVb: 30±2°C/75±5% RH.
- Number of Submission of Batches: Data from three pilot scale batches must be submitted and registered as a drug product.
- Stability Data: A minimum of 12 months of stability data is mandatory for drug products, per the ASEAN stability guidelines, when registering in ASEAN countries.
- CPP: Certificate of Pharmaceutical Product(9)
LATAM countries:
Due to the high differences between the countries of LATAM, these are the main differences and similarities between these countries:(10)
- Similarities:
- Quality, Safety, and Efficacy Standards for NCEs: Generally, Latin American National Regulatory Authorities (NRA) have harmonized requirements for the quality, safety, and efficacy of new chemical entities (NCEs). These requirements cover product manufacturing, characterization, stability testing, and clinical trials.
- Rigorous Regulatory Oversight: Across LATAM countries, there is a shared commitment to robust regulatory oversight to ensure the safety and efficacy of pharmaceutical products.
- Differences:
- Marketing Application Structure: Only Brazil and Cuba mandate that marketing applications adhere to the International Council for Harmonisation (ICH) Common Technical Document (CTD) structure. This standardized format is not a requirement in other LATAM countries, which may have their own submission formats.
- Definition of Generic Products: The definition of a generic product varies among LATAM countries:
- In Brazil and Mexico, “generic” refers to off-patent products marketed using the International Nonproprietary Name (INN) and proven to be interchangeable with the reference product (bioequivalent).
- In other countries like Argentina, Chile, Colombia, and Cuba, the term “generic” applies to off-patent products using the INN, irrespective of whether they have demonstrated bioequivalence.
- Regulatory Variations: LATAM countries may have individual regulatory nuances and specific requirements, contributing to a complex regulatory landscape for companies seeking NCE approvals across the region.
- Language and Documentation: Regulatory submissions may need to be in the official language of the respective country, Spanish, Portuguese, or another language, adding complexity to the submission process.
- Timelines and Review Processes: There can be variations in the timeframes and review processes for regulatory approvals among LATAM countries, impacting the speed at which products can enter the market.
In Latin American National Regulatory Authority (NRA) countries, there are policies regarding the renewal of marketing authorizations after a specified period. Typically, this renewal period is set at five years, notably different from regulators like the FDA in the United States and Health Canada, where marketing authorizations are issued indefinitely.
To ensure the continued validity of marketing authorizations, marketing authorization holders (MAHs) in Latin American NRAr countries must submit renewal requests well in advance. In most cases, these renewal requests must be submitted at least three months before the expiration date of the marketing authorization. The marketing authorization is automatically extended while the NRAr evaluates the request.
- Regulatory reliance:(10)
Regulatory reliance is a strategy National Regulatory Authorities (NRAs) employ to enhance the efficiency of their marketing application review processes. NRAs leverage regulatory reliance to expedite marketing authorizations for specific products or situations, which can be classified into two primary categories:
- Verification Approval: In this scenario, the NRA of the importing country permits a product to be marketed locally after confirming that it has already received authorization in the same form from one or more recognized reference authorities. Essentially, the NRA relies on the assessments and approvals made by these reference authorities, streamlining the process for local market entry.
- Abridged Approval: Under this approach, the NRA conducts a condensed and independent review that excludes certain elements. This abridged review is designed to accelerate the authorization process, making it more efficient for specific products or situations.
- Submission Requirements for Dossiers to Regulatory Bodies(11)
- Dossiers must be presented in the local language.
- A Certificate of Pharmaceutical Product (CPP) and Good Manufacturing Practices (GMP) manufacturing license are necessary.
- A Free Sale Certificate is required.
- Submission should include a Letter of Authorization or Power of Attorney.
- The embassy must legalize administrative documents.
- An API (Active Pharmaceutical Ingredient) Technical package is mandatory for Brazil and Mexico.
- Vendors must provide certificates of Analysis (COA) for API and Excipients.
- Detailed manufacturing procedures and controls should be included.
- Executed Batch Manufacturing Records and a Batch Numbering System are essential to the submission.
Gulf countries:
The Gulf Cooperation Council (GCC) is a coalition of six Arab nations in the Gulf region, united by shared economic and social objectives. These member countries consist of Bahrain, Kuwait, Oman, Qatar, Saudi Arabia, and the United Arab Emirates (UAE) and are collectively referred to as the GCC states.
One significant measure of harmonization adopted in the GCC area is adopting the eCTD (electronic Common Technical Document) format for submissions. As an illustration, Saudi Arabia initiated using the eCTD format in 2015 and has implemented it for different regulatory events such as renewal and various types of variation.(12)
PLG’s presence and support
We recognize that more and more pharma companies are learning about the importance of these international markets and having access to medicines. At PLG, we have established resources to support our clients in regulated and international markets. The global positioning of our offices allows us to accomplish these feats. As we continue to scale, we aim to enhance our existing services within these international markets to provide full-service coverage throughout the product life cycle.
Trust PLG to support your entire product portfolio in regulated and international markets.
References:
1: Regulatory requirement of ROW countries Blog: https://www.pharmaspecialists.com/2023/06/regulatory-requirements-of-row-countries.html#gsc.tab=0
2: Chapter 6 – Regulatory requirements of regulated market
https://www.sciencedirect.com/science/article/abs/pii/B9780128222119000046
3: Chapter 7 – Pharmaceutical regulatory requirements of nonregulated markets
https://www.sciencedirect.com/science/article/abs/pii/B9780128222119000022
4: The African Medicines Regulatory HarmonizationInitiative: Progress to Date https://esmed.org/MRA/index.php/mra/article/view/1668/1748
5: African medicines regulatory harmonization initiatives https://pdf.usaid.gov/pdf_docs/PA00TPBN.pdf
6: The Drug Regulatory Landscape in the ASEAN Region
6: ASEAN COMMON TECHNICAL REQUIREMENTShttps://asean.org/wp-content/uploads/2017/03/67.-December-2016-ACTR.pdf
7: COMPARATIVE STUDY OF REGULATORY REQUIREMENTS FOR THE COMPILATION AND APPROVAL OF DOSSIER; AS PER CTD, ACTD AND ECTD FORMATS https://www.ijpcbs.com/articles/comparative-study-of-regulatory-requirementsfor-the-compilation-and-approval-of-dossieras-per-ctd-actd-and-ectd-formats.pdf
8: REGULATORY REQUIREMENTS FOR THE REGISTRATION OF DRUGS IN ASEAN COUNTRIES https://wjpr.s3.ap-south-1.amazonaws.com/article_issue/c51f5be3662222c522bf436bfcdd4fd3.pdf
9: OVERVIEW OF DRUG REGISTRATION REQUIREMENTS FOR PHARMACEUTICALS IN EMERGING MARKET https://pdfs.semanticscholar.org/9583/5877b696619c51e07f97fcb0b8c84d64976b.pdf
10: REGULATORY SYSTEM STRENGTHENING IN THE AMERICAS LESSONS LEARNED FROM THE NATIONAL REGULATORY AUTHORITIES OF REGIONAL REFERENCE https://iris.paho.org/bitstream/handle/10665.2/53793/9789275123447_eng.pdf?sequence=5
11: Latin American countries Regulatory requirements overview https://pharmexcil.com/docs/Latin_American_Countries_Regulatory_requirements_30092011.pdf
12: Baseline eCTD Submission Requirements
Register to our news and events
Go to our Events to register
Go to our News to get insights
